市场监管总局关于发布《茶叶中美术绿(铅铬绿)的测定》食品补充检验方法的公告
市场监管总局关于发布
《茶叶中美术绿(铅铬绿)的测定》
食品补充检验方法的公告
(2019年第21号)
《茶叶中美术绿(铅铬绿)的测定》
食品补充检验方法的公告
(2019年第21号)
按照《食品补充检验方法工作规定》有关要求,《茶叶中美术绿(铅铬绿)的测定》食品补充检验方法已经国家市场监督管理总局批准,现予发布。
特此公告。
附件:茶叶中美术绿(铅铬绿)的测定.doc
市场监督管理总局
2019年5月13日
2019年5月13日
茶叶中美术绿(铅铬绿)的测定
BJS 201910
1 范围
本标准第一篇规定了茶叶中铬酸铅含量的测定方法。
本标准第一篇适用于茶叶中铬酸铅的测定。
本标准第二篇规定了茶叶中美术绿的定性方法。
本标准第二篇适用于茶叶中美术绿的定性判定。
2术语和定义
下列术语和定义适用于本标准。
2.1 美术绿
美术绿又名铅铬绿,用铬酸铅颜料沉淀到铁蓝颜料分散体上或使用铬酸铅颜料与铁蓝颜料混合均能制备出的一种颜料。美术绿的主要成分为铬酸铅和铁蓝,且铬酸铅的比例不低于50%。
第一篇铬酸铅的测定
铅含量的测定采用现行有效的国家标准GB 5009.12方法,CrO42-的测定选用下述方法。
第一法 高效液相色谱-电感耦合等离子体质谱联用法(HPLC-ICP/MS)
3 原理
茶叶样品经灰化后,其中的CrO42-经碱性提取液提取后,以液相色谱进行分离,分离后的目标化合物经过雾化由载气送入ICP炬焰中,经蒸发、解离、原子化、电离等过程,大部分转化为带正电荷的正离子,经离子采集系统进入质谱仪,质谱仪根据质荷比进行分离测定。以保留时间和质荷比定性,外标法定量。
4 试剂和材料
除非另有说明,本方法所用试剂均为优级纯,水为 GB/T 6682规定的一级水。
4.1 试剂
4.1.1硝酸(HNO3)。
4.1.2氨水(NH3·H2O)。
4.1.3氢氧化钠(NaOH):分析纯。
4.1.4无水碳酸钠(Na2CO3)。
4.2 试剂配制
4.2.1碱性提取液(0.1 mol/L NaOH和0.056 mol/L Na2CO3):称取4.0 ±0.05 g NaOH(4.1.3)和6.0±0.05g Na2CO3(4.1.4)溶解并定容至1 L,将溶液在20-25 ℃下密封保存于聚乙烯瓶中,使用时pH必须达到11.5以上,有效期1个月。
4.2.2碱性提取液(0.2 mol/L NaOH和0.112 mol/L Na2CO3):称取8.0 ±0.05 g NaOH(4.1.3)和12.0±0.05g Na2CO3(4.1.4)溶解并定容至1 L,将溶液在20-25 ℃下密封保存于聚乙烯瓶中,使用时pH必须达到11.5以上,有效期1个月。
4.2.3硝酸溶液(5 mol/L):取357 mL硝酸(4.1.1)用水定容至1000 mL,混匀待用。溶液在20-25 ℃下避光保存,如溶液呈黄色需重新配制。
4.2.4硝酸溶液(1+9):取10 mL硝酸(4.1.1)与90 mL水混合,摇匀。
4.2.5氨水溶液(1+9):取10 mL氨水(4.1.2)与90 mL水混合,摇匀。
4.2.6 硝酸铵溶液(0.05 mmol/L):一定量水中加入3.4 mL硝酸(4.1.1)和3.7 mL氨水(4.1.2),用水定容至1 L,摇匀;用硝酸溶液(4.2.4)或氨水溶液(4.2.5)调节pH在7.0-7.2范围内。
4.3 标准品
铬酸铅:Lead chromate,CAS号:7758-97-6,分子式PbCrO4,分子量323.18,纯度≥99%。
4.4 标准溶液的配制
4.4.1 CrO42-标准储备液:准确称取0.0279 g铬酸铅标准物质,于100 mL烧杯中,加入50 mL碱性提取液(4.2.2)置于恒温磁力搅拌水浴锅中,在90℃-95 ℃温度下不间断搅拌30 min,取出后冷却至室温后转移至100 mL容量瓶中,用水冲洗烧杯并转移至容量瓶中,用水定容至刻度。溶液中CrO42-的浓度为100 mg/L。或使用六价铬标准溶液进行配制。
4.4.2 CrO42-标准工作液:
取1.0 mLCrO42-标准储备液(4.4.1)用硝酸铵溶液(4.2.6)定容至10 mL,制得标准中间液,浓度为10 mg/L。
分别吸取0.05,0.1,0.2,0.5,1.0,2.0,5.0 mL标准中间液于100 mL容量瓶中,用硝酸铵溶液(4.2.6)定容并混匀,过水系微孔滤膜(0.45 μm),待上机测定。该标准工作液CrO42-的浓度分别为5 μg/L,10 μg/L,20 μg/L,50 μg/L,100 μg/L,200 μg/L,500 μg/L。该标准工作液现用现配。
5 仪器和设备
注:玻璃器皿及坩埚均需以硝酸溶液(1+4)浸泡2 h,用水反复冲洗,最后用去离子水冲洗干净。
5.1 高效液相色谱-电感耦合等离子体质谱联用仪(HPLC-ICP/MS)。
5.2 分析天平:感量为0.1 mg和0.01 g。
5.3 可调式电阻炉,功率为2000 W。
5.4 磁力搅拌恒温水浴锅:能在90 ℃-95 ℃恒温。
5.5 pH计:精度为0.01。
5.6 样品粉碎装置。
6 试样制备与保存
茶叶样品去杂后,放入粉碎机中粉碎,样品全部过标准网筛(425 μm),储于洁净容器中,至于阴凉干燥处密封保存,备用。
7 分析步骤
7.1 样品提取
准确称取粉碎均匀的茶叶样品1 g(精确到0.01 g)于100 mL坩埚中,将坩埚置于电炉上以最大功率灼烧至无白烟。冷却至室温后加入50 mL碱性提取液(4.2.1),置于磁力搅拌恒温水浴锅中,在90 ℃-95 ℃温度下不间断搅拌30 min,取出后冷却至室温,定容至100 mL容量瓶中,从中取1.0 mL用硝酸铵溶液(4.2.6)定容至50 mL(或根据实际浓度适当稀释),过水系微孔滤膜(0.45 μm),待上机测定。按同一操作方法作空白试验。
7.2 仪器参考条件
7.2.1 液相色谱参考条件
色谱柱:G3268-80001 色谱柱,(柱长30 mm,内径4.6 mm),或等效柱。
流动相:0.05 mmol/L的硝酸铵溶液(4.2.6)。
洗脱方式:等度洗脱。
流速:0.6 mL/min。
进样量:50 μL。
7.2.2 电感耦合等离子体质谱仪参考条件
模式:碰撞反应池模式;RF入射功率为1600 W;载气为高纯氩气;载气流速为1.0 L/min;雾化器:漩流雾化器;碰撞反应气(He),流速为3.5 mL/min;检测质量数m/z=52(Cr)。
7.3 定量测定
7.3.1 标准曲线的制作
吸取标准系列溶液(4.4.2)50 μL注入液相色谱-电感耦合等离子质谱仪,按仪器参考条件(7.2)进行测定,得到相应的色谱图,以保留时间定性。以标准系列溶液中目标化合物的浓度为横坐标,以色谱峰面积为纵坐标,绘制标准曲线。
7.3.2 试样溶液的测定
在相同条件下,吸取空白溶液、试样溶液(7.1)50 μL注入液相色谱-电感耦合等离子质谱联用仪进行测定,得到相应色谱图(参见附录A),以保留时间定性。根据标准曲线得到待测液中组分的含量,平行测定次数不少于两次。
8 分析结果表述
8.1试样中铬酸根(CrO42-)的含量按式(1)计算:
……………………(1)
式中:
X ——试样中CrO42-的含量,单位为毫克每千克(mg/kg);
C1 ——样品溶液中CrO42-的浓度,单位为微克每升(μg/L);
C0 ——溶剂空白中CrO42-的浓度,单位为微克每升(μg/L);
V ——试样溶液定容体积,单位为毫升(mL);
ƒ——试样溶液稀释倍数;
1000——换算系数;
m ——样品质量,单位为克(g)。
8.2试样中铬酸铅含量的计算:
当CrO42-含量Pb含量≥0.5597时,试样测得的Pb含量乘以换算系数1.560,即得铬酸铅(以PbCrO4计)的含量;当CrO42-含量/Pb含量<0.5597时,试样测得的CrO42-含量乘以换算系数2.787,即得铬酸铅(以PbCrO4计)的含量。
计算结果保留三位有效数字。
9 检测方法的精密度、灵敏度、准确度
9.1 精密度
在重复条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的10%。
9.2 灵敏度
当称样量为1.0 g,定容体积为100 mL后稀释50倍测定时,PbCrO4检出限为10 mg/kg,定量限30 mg/kg。
9.3 准确度
本方法CrO42-添加浓度在30~300 mg/kg范围内,回收率为87.1%~105.9%。
第二法 离子色谱法(IC法)
10 原理
茶叶样品经灼烧后,其中的CrO42-经碱性提取液提取,在碱性条件下(pH=9.0±0.5),采用阴离子交换色谱柱分离,离子色谱-电导检测器测定,外标法定量。
11试剂和材料
除非另有说明,本方法所用试剂均为优级纯,水为 GB/T 6682规定的一级水。
11.1 试剂
11.1.1 浓硫酸(H2SO4)。
11.1.2 硝酸(HNO3)。
11.1.3 碳酸氢钠(NaHCO3)。
11.1.4 氢氧化钠(NaOH):分析纯。
11.1.5无水碳酸钠(Na2CO3)。
11.2试剂配制
11.2.1 碱性提取液:配制同4.2.1。
11.2.2 硝酸溶液(5 mol/L):配制同4.2.3。
11.2.3 淋洗液储备液(40 mmol/L碳酸钠和10 mmol/L碳酸氢钠混合溶液):分别称取4.25 g无水碳酸钠(11.1.5)和0.84 g碳酸氢钠(11.1.3),用水溶解并定容至1 L,混匀待用。
11.2.4 淋洗液使用液(4 mmol/L碳酸钠和1 mmol/L碳酸氢钠混合溶液):将淋洗液储备液(11.2.3)用水稀释10倍,混匀,经水系微孔滤膜(0.45 μm)抽滤后使用。
11.2.5 再生液:吸取2 mL浓硫酸(11.1.1)用水定容至1L,摇匀,经水系微孔滤膜(0.45 μm)抽滤后使用。
11.3 标准品
同4.3。
11.4 标准溶液的配制
11.4.1铬酸根(CrO42-)标准储备液:配制同4.4.1。
11.4.2 CrO42-标准工作液:
取浓度为100 mg/L的CrO42-标准储备液(4.4.1)10 mL用水定容至100 mL,制得标准中间液,浓度为10 mg/L。
分别吸取0.2,0.5,1.0,5.0,10.0,20.0 mL标准中间液于100 mL容量瓶中,用水定容并混匀,过水系微孔滤膜,待上机测定。该标准工作溶液CrO42-的浓度分别为0.02 μg/mL,0.05 μg/mL,0.10 μg/mL,0.50 μg/mL,1.00 μg/mL,2.00 μg/mL。该标准工作液现用现配。
12 仪器和设备
12.1 离子色谱仪,配电导检测器,碳酸盐淋洗系统。如采用氢氧根系统,条件见附录C。
12.2 分析天平:感量0.1 mg和0.01 g。
12.3 可调式电阻炉,功率为2000 W。
12.4 磁力搅拌恒温水浴锅:能在90 ℃-95 ℃恒温。
12.5 pH计:精度为0.01。
12.6 样品粉碎装置。
13 试样制备与保存
同6。
14 分析步骤
14.1 样品提取
准确称取粉碎均匀的茶叶样品1 g(精确到0.01 g)于100 mL坩埚中,将坩埚置于电炉上以最大功率灼烧至无白烟。冷却至室温后加入50 mL碱性提取液(4.2.1),置于恒温磁力搅拌水浴锅中,在90 ℃-95 ℃温度下不间断搅拌30 min,取出后冷却至室温,转移至100 mL烧杯中,用硝酸溶液(4.2.3)调pH至9.0±0.5后转移至500 mL容量瓶中,用水定容并混匀。过水系微孔滤膜(0.45 μm),待上机测定。按同一操作方法作空白试验。
14.2 离子色谱条件
色谱柱:Metrosep A Supp 5-150或相当的阴离子色谱柱。
检测器:电导检测器。
抑制器:MSM ǁ 化学抑制器。
柱温:30 ℃。
流速:0.7 mL/min。
进样量:20 μL。
14.3 定量测定
14.3.1 标准曲线的制作
吸取标准系列溶液(11.4.2)20 μL注入离子色谱仪,按仪器参考条件(14.2)进行测定,得到相应的色谱图,以保留时间定性。以标准系列溶液中目标化合物的浓度为横坐标,以色谱峰的峰面积为纵坐标,绘制标准曲线。
14.3.2 样品测定
在相同条件下,分别吸取空白溶液、试样溶液(14.1)20 μL,按仪器参考条件(14.2)进行测定,得到相应色谱图(参见附录B),以保留时间定性。根据标准曲线得到待测液中组分的浓度,平行测定次数不少于两次。
15 结果计算
15.1试样中铬酸根(CrO42-)的含量按式(2)计算:
……………(2)
式中:
X ——试样中CrO42-的含量,单位为毫克每千克(mg/kg);
C1 ——样品溶液中CrO42-的浓度,单位为微克每毫升(μg/mL);
C0 ——样品空白液中CrO42-的浓度,单位为微克每毫升(μg/mL);
V ——试样溶液定容体积,单位为毫升(mL);
1000——换算系数;
m ——样品质量,单位为克(g)。
15.2试样中铬酸铅含量的计算:
同8.2。
计算结果保留三位有效数字。
16 方法的精密度、灵敏度、准确度
16.1 精密度
在重复条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的10%。
16.2 灵敏度
当称样量为1.0 g,定容体积为500 mL时,PbCrO4检出限为10 mg/kg,定量限30 mg/kg。
16.3 准确度
本方法CrO42-添加浓度在30~300 mg/kg范围内,回收率为85.17~107.59%。
第二篇 铁蓝定性验证
17 原理
铁蓝的主要成分亚铁氰根在酸性条件下蒸馏分解出氰化物,与氯胺T反应,生成氯化氰,与显色剂生成蓝色染料,分光光度法测定。
18 试剂和材料
除非另有说明,本方法所用试剂均为分析纯,水为 GB/T 6682规定的三级水。
18.1 试剂
18.1.1 氢氧化钠(NaOH)。
18.1.2 无水磷酸氢二钠(Na2HPO4)。
18.1.3 无水磷酸二氢钾(KH2PO4)。
18.1.4 氯胺T(C7H7ClNNaO2S·3H2O)。
18.1.5 酒石酸(C4H6O6)。
18.1.6 酚酞(C20H14O4)。
18.1.7 1-苯基-3-甲基-5-吡唑啉酮(C10H10N2O)。
18.1.8 冰乙酸(CH3COOH)。
18.1.9 无水乙醇(CH3CH2OH)。
18.1.10 吡啶(C5H5N)。
18.1.11 甲基橙(C14H14N3SO3Na)。
18.2试剂配制
18.2.1 NaOH溶液(0.1 mol/L):准确称取1.0 g NaOH(18.1.1) ,用水定容至250 mL,摇匀待用。
18.2.2 NaOH溶液(0.5 mol/L):准确称取5.0 g NaOH(18.1.1) ,用水定容至250 mL,摇匀待用。
18.2.3磷酸盐缓冲溶液(pH=7.0):准确称取42.6 g无水磷酸氢二钠(18.1.2)和13.6 g无水磷酸二氢钾(18.1.3),加水溶解,定容至1L,摇匀待用。
18.2.4氯胺T溶液(10 g/ L):准确称取 1.0 g氯胺T(18.1.4),用水定容至100 mL,摇匀待用,现用现配。
18.2.5酚酞指示液:准确称取1.0 g酚酞(18.1.6)于60 mL 无水乙醇(18.1.9)中,加水40mL,摇匀待用。
18.2.6吡啶-吡唑啉酮溶液(5 g/L):准确称取1.0 g 1-苯基-3-甲基-5-吡唑啉酮(18.1.7),溶于200 mL无水乙醇(18.1.9)中,加1.2 mL吡啶(18.1.10),混匀待用,现用现配。
18.2.7乙酸溶液(0.1 mol/ L):准确吸取1.15 mL冰乙酸(18.1.8),用水定容至200 mL,摇匀待用。
18.2.8甲基橙指示剂(0.5 g/ L):准确称取50 mg甲基橙(18.1.11),溶于水中,定容至100 mL,摇匀待用。
18.3 标准品
铁蓝:Prussion Blue,CAS号:14038-43-8,分子式Fe4[Fe(CN)6]3,分子量859.23,纯度≥99%。
18.4参比溶液配制
准确称取0.0150 g铁蓝标准物质于250 mL高型烧杯中,准确加入100 mL NaOH溶液(18.2.1),于室温下震荡30 min后,取10.0 mL定容至100 mL,取稀释液1.0 mL于500 mL蒸馏烧瓶中,加水至液体总量约为150 mL,加入1~2滴甲基橙指示剂(18.2.8),再加入2 g酒石酸(18.1.5),溶液由橙黄色变为橙红色,迅速连接好蒸馏装置,将冷凝管下端插入盛有10 mL NaOH溶液(18.2.2)的100 mL容量瓶中,收集蒸馏液,调节温度将蒸馏速度控制在2 mL/min~3mL/min,当蒸馏液接近100 mL时,停止加热,取下容量瓶,用水定容至刻度,摇匀。此标准溶液中铁蓝的浓度为0.15 mg/L。
19 仪器和设备
19.1 分光光度计:配1 cm比色池。
19.2 分析天平:感量0.01 mg和0.01 g。
19.3 500 mL蒸馏装置:配冷凝器和聚乙烯毛细管。
19.4 振荡器。
19.5 离心机。
20分析步骤
20.1试样制备
准确称取未经粉碎的茶叶样品10 g(精确至0.01 g),准确加入100 mL NaOH溶液(18.2.1)于250 mL 高型烧杯中,于室温下震荡30 min后,用漏斗滤去茶叶,滤液于3000 r/min离心5 min,准确移取上清液50 mL于500 mL蒸馏烧瓶中,加水至液体总量约为150 mL,加入1~2滴甲基橙指示剂(18.2.8),再加入2 g酒石酸(18.1.5),溶液由橙黄色变为橙红色,迅速连接好蒸馏装置,将冷凝管下端插入盛有10 mL NaOH溶液(18.2.2)的 100 mL容量瓶中,收集蒸馏液,调节温度将蒸馏速度控制在2 mL/min~3 mL/min,当蒸馏液接近100 mL时,停止加热,取下容量瓶,用水定容至刻度,摇匀,待测。同时作空白试验,空白溶液除不加碱液(18.2.1)和酒石酸(18.1.5)外,均按(20.1)操作。
20.2 测定
准确移取参比溶液(18.4)、样液(20.1)、空白溶液(20.1)及水(仪器零点校正液)各5.0 mL置于25 mL比色管中,加入1滴酚酞指示液(18.2.5),用乙酸溶液(18.2.7)缓慢调至无色,加4.0 mL磷酸缓冲溶液(18.2.3)和0.3 mL氯胺T溶液(18.2.4),摇匀,静置3 min,加5.0 mL吡啶-吡唑啉酮溶液(18.2.6),摇匀,静置20 min,加水至刻度,摇匀。分光光度计于620 nm处,用1 cm比色池,测定吸光度值后比较。
21 定性判定
当扣除空白的样液吸光度值≥参比溶液的吸光度值时,即铁蓝有检出,反之,铁蓝未检出。
22 方法的定量限
当称样量为10.0 g,蒸馏烧瓶移取量为50 mL,定容体积为100 mL时,铁蓝的定量限为3 mg/kg。
23 美术绿综合判定
本方法通过测定茶叶中的铅和铬酸根,判定茶叶中是否含有铬酸铅。当铬酸铅检出时,需要定性测定茶叶中铁蓝,当铁蓝检出时,判定茶叶中含有美术绿。茶叶中的美术绿以铬酸铅的含量表示,根据8.2计算铬酸铅的含量,从而表示茶叶中美术绿的含量。
附 录 A
HPLC-ICP/MS法的色谱图
A.1 标准溶液色谱图(HPLC-ICP/MS法)见图A.1。
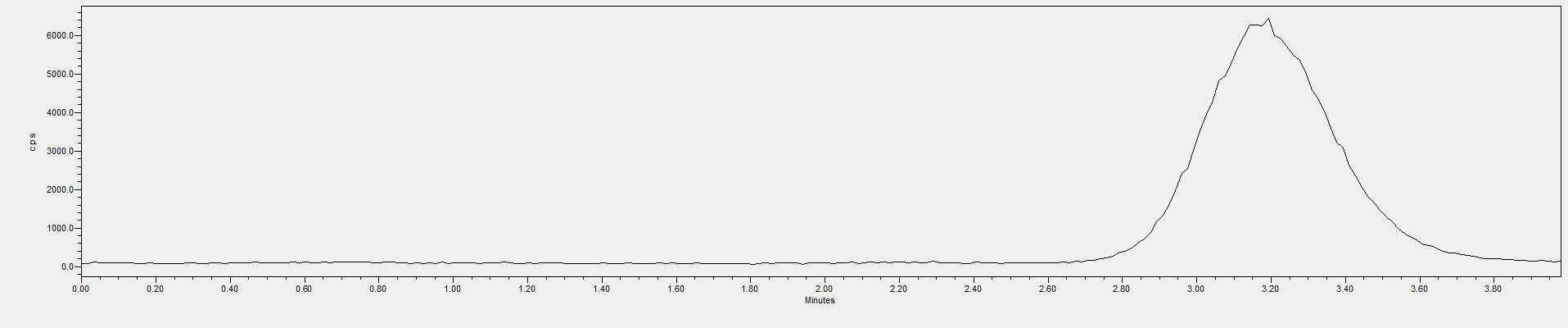
图A1 HPLC-ICP/MS测得CrO42-标准溶液色谱图(40 μg/L)
A.2 基质加标色谱图(HPLC-ICP/MS法)见图A.2。
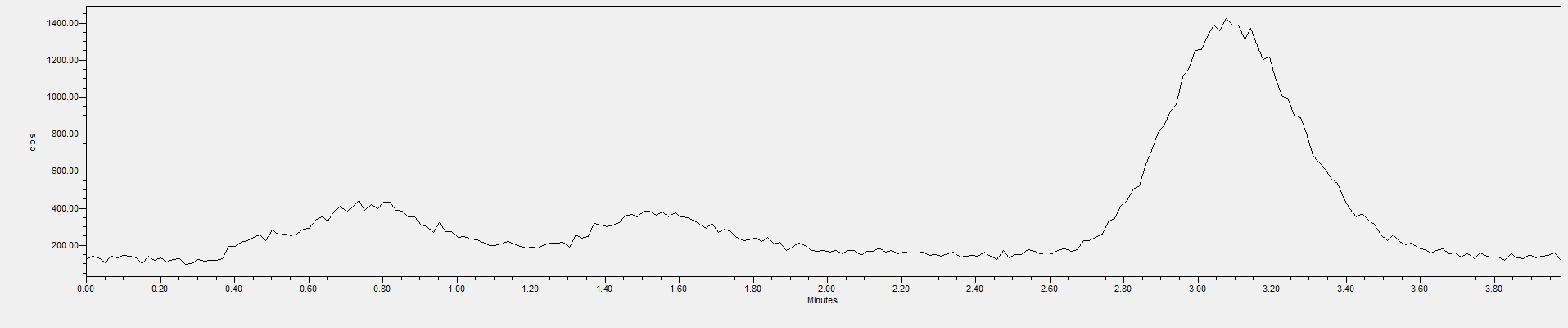
图A.2 HPLC-ICP/MS测得基质加标样品测定液中的CrO42-色谱图(20 μg/L)
附 录 B
离子色谱法的色谱图
B.1 标准溶液色谱图(离子色谱法)见图B.1。
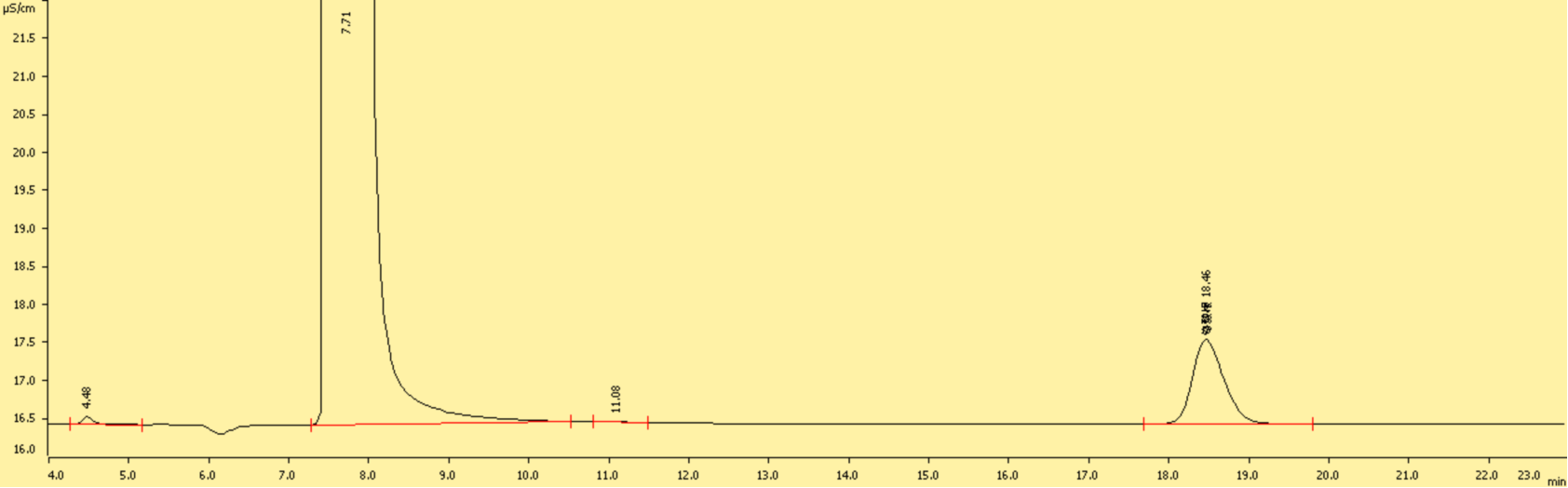
图B1 离子色谱测得CrO42-标准溶液色谱图(10 mg/L)
B.2 基质加标色谱图(离子色谱法)见图B.2。
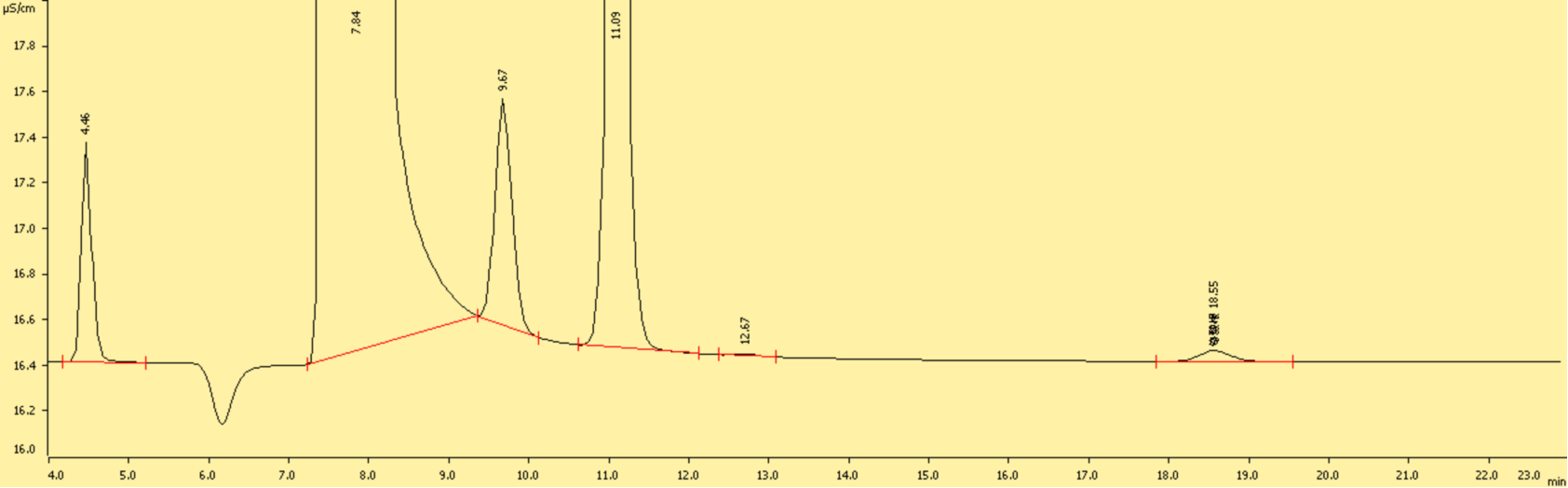
图B2 离子色谱测得基质加标样品测定液中的CrO42-色谱图(0.2 mg/L)
本方法负责起草单位:吉林省食品检验所
方法的参与验证单位:大连市食品检验所、广西-东盟食品药品安全检验检测中心、海南省食品检验检测中心、辽宁省食品检验检测院、北京市食品质量监督检验三站。
主要起草人:石金娥、华蕾、王莹、李滢倩、王庆峰、刘斌。
